By Dr. Khadiga Elfadil Ahmed Mohammed
Abstract:
Background:
Rosai-Dorfman syndrome (RDS) is a rare, benign histiocytic
disorder usually presenting with cervical lymphadenopathy but
often affecting extranodal sites including the paranasal sinuses,
orbit, and central nervous system. Treatment is not
standardized. While some patients improve spontaneously or
with corticosteroids, others require surgery, systemic therapy,
or radiotherapy.
Case Presentation:
A 24-year-old man with biopsy-confirmed RDS presented with
temporal and sphenoid sinus disease, bilateral cervical
lymphadenopathy, and right optic nerve compression causing
severe visual loss (6/60). His disease, first diagnosed in 2018,
had progressed despite repeated steroid therapy. Surgery was
considered high-risk, and the response to systemic treatment
was modest especially in the setting of intracranial involvement. Given the threat to vision, radiotherapy was
chosen. He was treated with 45 Gy in 25 fractions (Figure 1)
The course required adaptive replanning because of soft tissue
and nodal changes.
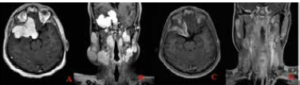
Fig 1) A-B) Axial and coronal T1 and post-contrast images at presentation
revealing the extent of disease, C-D) 12 months post-radiotherapy showing
further interval reduction in size and extent of widespread dural thickening,
with decreased extension, reduced enhancement, and improved mass effect. Follow-up imaging at four months post radiotherapy showed
partial regression of intracranial and cervical disease. At one
year, further significant reduction was seen. Vision remained
stable without further decline. No major late toxicities were
reported.
Conclusion:
This case demonstrates that radiotherapy can provide effective
and durable disease control in RDS with critical organ
involvement. Although rarely used, it should be considered
when other options are unsuitable or ineffective.